PaDEL-Descriptor
Description
A software to calculate molecular descriptors and fingerprints. The software currently calculates 1875 descriptors (1444 1D, 2D descriptors and 431 3D descriptors) and 12 types of fingerprints (total 16092 bits). The descriptors and fingerprints are calculated using The Chemistry Development Kit with additional descriptors and fingerprints such as atom type electrotopological state descriptors, Crippen's logP and MR, extended topochemical atom (ETA) descriptors, McGowan volume, molecular linear free energy relation descriptors, ring counts, count of chemical substructures identified by Laggner, and binary fingerprints and count of chemical substructures identified by Klekota and Roth.
Screenshot
Requirements
Java JRE version 6 and above is recommended.
Usage
Graphical user interface
Launch the software from here (recommended but requires Java Web Start) or download it from here. If you download the zipped file, unzip the files to any directory and launch the software using "java -jar PaDEL-Descriptor.jar" without the double quotes.
Select a single structural file or a directory containing the molecules' structural files. Most common file formats (e.g. MDL mol, SMILES) are supported but the recommended file format is MDL mol.
Select a file to save the calculated descriptors to. The descriptors will be saved in comma separated value (CSV) file format. The first row is the header row. Subsequent rows will contain the calculated descriptors for one molecule per row. The first column is the molecule's name, which is either obtained from the structural file or autogenerated (will be prefixed with AUTOGEN_ followed by the file name). Subsequent columns are the descriptors for the molecules.
Check the option "1D & 2D" if you wish to calculate 1D and 2D descriptors.
Check the option "3D" if you wish to calculate 3D descriptors.
Check the option "Fingerprints" if you wish to calculate fingerprints.
Check the option "Remove salt" if you wish to remove salts like Na, Cl from the molecule before calculation of descriptors. It is better to remove salts from the molecule using your own means than to rely on this software to do the job.
Check the option "Detect aromaticity" if you wish to remove existing aromaticity information and automatically detect aromaticity in the molecule before calculation of descriptors. This option was implicitly checked in versions prior to 2.8 and was not working properly prior to 2.12. Note that this will remove any 3D information from the molecules unless the "Retain 3D coordinates" option is enabled. However, retaining 3D coordinates might prevent this from working properly.
Check the option "Standardize tautomers" if you wish to standardize tautomers. Note that this will remove any 3D information from the molecules unless the "Retain 3D coordinates" option is enabled. However, retaining 3D coordinates might prevent this from working properly.
Select a SMIRKS tautomer file if you wish to standardize tautomers using SMIRKS. An example of such a file can be downloaded from here. If you did not specify any file, the default file found in META-INF in the jar file will be used.
Check the option "Standardize nitro groups" if you wish to standardize nitro groups to N(:O):O. This is necessary for extended topochemical atom (ETA) descriptors to be calculated correctly.
Check the option "Retain 3D coordinates" if you wish to retain 3D coordinates of the molecules when "Detect aromaticity" and/or "Standardize tautomers" are enabled. However, retaining 3D coordinates might prevent the detection of aromaticity and standardization of tautomers from working properly.
Check the option "Convert to 3D" if you wish to convert the molecule to 3D before calculation of descriptors. It is better to convert the molecule to 3D using your own means than to rely on this software to do the job.
Check the option "Log" if you wish to generate a log file.
Enter a value (greater than 0) for the option "Max. threads" if you wish to limit the maximum number of threads to use. By default (any values less than 0), PaDEL-Descriptor will use as many threads as the number of cpu cores available.
Enter a value (greater than 0) for the option "Max. waiting jobs" to set the maximum number of jobs to store in queue for worker threads to process. By default (any values less than 0), this is set as 50*Max threads. If this value is too high, more memory will be used and more processing time will be spent queuing jobs rather than doing them. If this value is too low, worker threads may end up waiting for jobs rather than doing them.
Enter a value (greater than 0) for the option "Max compounds per file" if you wish to limit the maximum number of compounds to be saved to a descriptor file. This is useful for limiting the size of a descriptor file and may help to prevent slowing down of descriptor calculation due to writing of new descriptor values to a large descriptor file.
Enter a value (greater than 0) for the option "Max. running time per molecule" if you wish to restrict the maximum descriptor calculation time (in milliseconds) for a molecule. Use -1 to remove restriction on running time.
Check the option "Retain molecules order" if you wish to retain order of molecules in structural files for descriptor file. Note that this may lead to large memory use if descriptor calculations are stuck at one molecule as the others will not be written to file and cleared from memory.
Check the option "Use filename as molecule name" if you wish to use the filename (minus the extension) as molecule name.
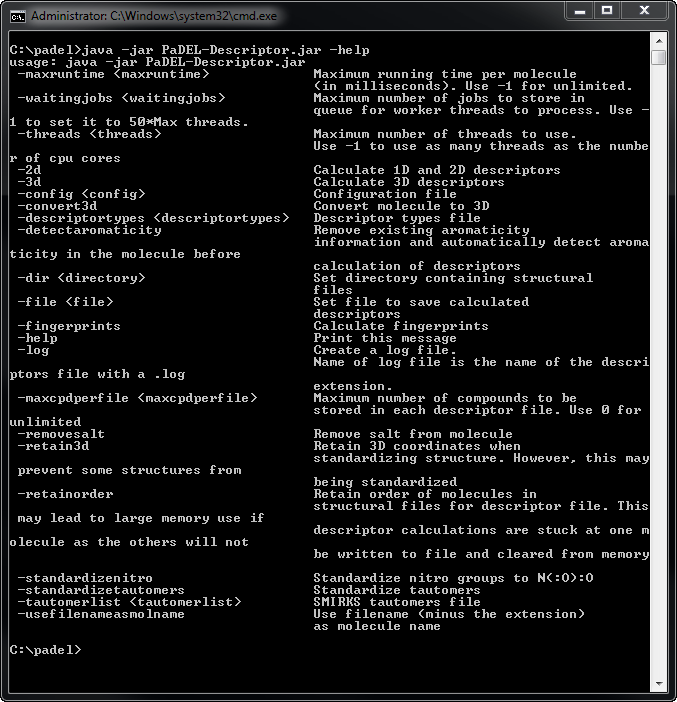
Command line
If you have downloaded the zipped file, you can use it without the graphical interface. Unzip the files to any directory and run the software using "java -jar PaDEL-Descriptor.jar -help" to view the various options that are available
RapidMiner extension
Installation
For RapidMiner v5.0
- Copy PaDEL-Descriptor.jar into <Installation Dir>\Rapid-I\RapidMiner5\lib\plugins. Copy all jar files in lib into <Installation Dir>\Rapid-I\RapidMiner5\lib.
For RapidMiner v5.1.6 and above
- Copy PaDEL-Descriptor.jar into <Installation Dir>\Rapid-I\RapidMiner5\lib\plugins. Copy all jar files in lib into <Installation Dir>\Rapid-I\RapidMiner5\lib\plugins\lib.
Usage
A new category "Chemistry" will be found in the Operators tab. To calculate descriptors, you can use the operator "Read Compounds" to read in a set of compounds and then join it to the operator "Calculate Descriptors" to calculate molecular descriptors for these compounds.
KNIME extension
Installation
Add "http://www.yapcwsoft.com/dd/padeldescriptor/knime" to the Available Software Sites in KNIME or download a zipped update site.
See here for a list of changes
Usage
A new category "PaDEL" will be found in the Node Repository tab. To calculate descriptors, you can use the operator "Compounds Reader" to read in a set of compounds and then join it to the node "PaDEL-Descriptor" to calculate molecular descriptors for these compounds.
Known limitations
It is not possible to use the CDK "Molecule to CDK" node as input to the "PaDEL-Descriptor" node as the CDK extension uses CDK 1.5.x whereas PaDEL-Descriptor uses CDK 1.4.x. Hence, there are some compatibility issues which will only be resolved when PaDEL-Descriptor updates to CDK 1.5.x, which will only happen when CDK 1.5.x becomes the new stable release.
Licence
This software is free for all (e.g. personal, academic, non-profit, non-commercial, government, commercial, etc) to use.
Citation
Please cite using: Yap CW (2011). PaDEL-Descriptor: An open source software to calculate molecular descriptors and fingerprints. Journal of Computational Chemistry. 32 (7): 1466-1474
Source code
Download the source code here. The source code is released as public domain. However, any part of the code that uses CDK, AMBIT2, RapidMiner, Apache Commons CLI, and l2fprod will still be restricted by the respective licenses.
Known Problems
- Folder selection window for "Molecules directory" may experience some initial lag when using on machines with Vista / Windows 7.
- SMILES files need to have a .smi extension in order to be detected as SMILES files.
- ALOGP, ALOGP2, AMR may not be accurate because the existing algorithm may not be able to assign all atoms in a molecule to an atom type defined by Viswanadhan et al.
- "Detect aromaticity" option should be unchecked for structural files where aromatic bonds have been explicitly defined. Otherwise, the automatic detection of aromaticity may override and remove the original aromatic bonds.
- For calculation of extended topochemical atom (ETA) descriptors, the following precautions should be taken unless the "Detect aromaticity" and "Standardize nitro groups" options are checked (thanks to A/Prof Kunal Roy for suggesting these precautions).
- Any heteroaromatic or carbocyclic aromatics system should be explicitly drawn with aromatic bonds.
- The nitro group when attached to any aromatic or heteroaromatic ring must be drawn with aromatic bonds. However, in case of nitro aliphatics, the nitro group may be drawn using double bonds.
- It is better to draw compounds with explicit hydrogens. This is mandatory for nitro group containing compounds and heteroaromatic compounds to get correct values for all ETA descriptors.
- "Detect aromaticity" and "Standardize tautomers" will remove 3D information from the molecules. This prevents 3D descriptors from being calculated if these two options are enabled. Using the "Retain 3D coordinates" option may prevent these two options from working properly. Thus for the calculation of 3D descriptors, it is best to specify aromaticity and standardize tautomers using your own means and disable these two options. For the calculation of 2D descriptors, it is best to enable these two options, together with "Standardize nitro groups" in order to ensure the same descriptor values are calculated for the same molecule stored in different file formats with different aromaticity information or different tautomers.
- "Standardize tautomers" may create strange tautomers for some compounds. Hence, it should be unchecked for now. This issue will be addressed in the subsequent versions.
- The option "Max. running time per molecule" will attempt to stop the descriptor calculation for a molecule if it exceeds the specified time. However, due to the nature of Java in handling threads, the descriptor calculation will not stop immediately for a molecule. This may cause an accumulation of threads running in the background. Hence, do not set this running time too short. 30000 to 60000 milliseconds should be sufficient for most molecules.
History
- Version 2.21 [21 Jul 2014]: Fixed bug involving descriptors based calculation of intrinsic states (e.g. electrotopological state, extended topochemical atom) that sometimes prevents the calculation of these descriptors. Modified the chi path descriptors such that average simple path and average valence path will output zero instead of blank if there are no paths of a particular order. Modified Barysz matrix, detour matrix and topological distance descriptors so that they will not given invalid values for EE and VE3 or very large values for VR1 and VR2.
- Version 2.20 [16 Jul 2014]: Fixed bug involving descriptors based on Barysz matrix and Burden modified eigenvalues that prevent program from stopping when molecule contained invalid atoms.
- Version 2.19 [26 Jun 2014]: Added Average molecular weight, 346 2D autocorrelation descriptors (these replaced the previous 15 autocorrelation descriptors and the values of ATSc1-c5, ATSm1-m5 and ATSp1-p5 will have different values from previous version), 91 descriptors based on Barysz matrix, 96 Burden modified eigenvalues, 16 chi path descriptors (average simple path and average valence path), 12 constitutional descriptors, 11 descriptors based on detour matrix, 42 information content descriptors, 22 path counts descriptors, 20 topological charge descriptors (globalTopoChargeIndex was shifted from topological descriptor to this and renamed as JGT), 11 descriptors based on topological distance matrix, 20 walk counts descriptors, 80 3D autocorrelation descriptors, 210 RDF descriptors and 91 WHIM descriptors (the previous 85 WHIM descriptors were removed as the weightings were not consistent with the new set of descriptors such as autocorrelation, Barysz, Burden modified, etc. G1 to G3 descriptors could not be calculated due to error in algorithm which could not be fixed). Add two fingerprints based on 2D atom pairs. Improve calculation speed for WeightedPathDescriptor, ElectrotopologicalStateAtomTypeDescriptor, ExtendedTopochemicalAtomDescriptor.
- Version 2.18 [01 Jul 2013]: Added 3 rotatable bond descriptors (RotBFrac, nRotBt, RotBtFrac), 34 ring count descriptors for rings containing heteroatoms, 3 topological descriptors (topoRadius, topoDiameter, globalTopoChargeIndex), 2 geometrical descriptors (geomRadius, geomDiameter). Shifted topoShape descriptor from PetitjeanShapeIndexDescriptor (3D descriptor) to TopologicalDescriptor (2D descriptor) since it is a 2D descriptor so as to prevent repeated calculations of distance matrix. Fixed bug in dearomatization of some compounds. Thanks to Dr Carol Marchant for reporting the bug.
- Version 2.17 [27 Mar 2013]: Fixed bug in Fragment complexity descriptor where it includes the number of bonds to hydrogen atoms in its calculation. Thanks to Dr Tom Dickson for reporting the bug.
- Version 2.16 [15 Mar 2013]: Fixed bug in Carbon types descriptors where it may calculate the number of carbons types wrong for aromatic compounds. Thanks to Prof Paola Gramatica and Dr Stefano Cassani for reporting the bug.
- Version 2.15 [04 Feb 2013]: Fixed bug introduced in v2.14 preventing SubstructureFingerprintCount and KlekotaRothFingerprintCount from working. Thanks to Daichi Yukihira from Kyushu University for reporting the bug.
- Version 2.14 [04 Dec 2012]: Bug fixes for mol2 files. Added option to restrict maximum descriptor calculation time for a molecule as suggested by Dr Filip Stefaniak. Developed KNIME nodes for PaDEL-Descriptor. Updated CDK library to 1.4.15.
- Version 2.13 [21 Sep 2012]: Update "Standardize nitro groups" to standarize nitro groups to N+(=O)O-, following a feedback by Prof Paola Gramatica and Dr Stefano Cassani. Updated "Detect aromaticity" code to handle dearomatization and re-aromatization of rings better and to dearomatize conjugation compounds. Due to these changes, some descriptors (e.g. nH, nBondsM, nHBDon_Lipinski, MAXDN2, MAXDP2 and DELS2) may have different values from v2.12, especially for compounds containing nitro groups, multiple fused aromatic rings and conjugation. Updated CDK library to 1.4.13.
- Version 2.12 [08 May 2012]: Added the following descriptors (nX, nBondsM, CrippenLogP, CrippenMR, MAXDN2, MAXDP2 and DELS2) as suggested by Prof Paola Gramatica and Dr Stefano Cassani. Fixed a bug in atom type electrotopological state which prevented some aromatic atoms from being recognized as hydrogen bond donors or acceptors. Updated "Detect aromaticity" code and added "Standardize tautomers" and "Standardize nitro groups" to help standardize molecules prior to 2D descriptors calculations. Updated CDK library to a customized version of 1.4.9, which fixed some concurrency problems. Uses AMBIT2 library to standardize tautomers and handling of SMIRKS. Replaced "Remove salt" and "Detect aromaticity" operators for RapidMiner with "Standardize" operator. Added "Get SMILES" operator for RapidMiner.
- Version 2.11 [30 Dec 2011]: Fixed a bug in extended topochemical atom (ETA) descriptors which did not take into account conjugation with an aromatic system. Thanks to A/Prof Kunal Roy for pointing it out.
- Version 2.10 [22 Dec 2011]: Added 43 extended topochemical atom (ETA) descriptors as suggested by the developer, A/Prof Kunal Roy. A/Prof Kunal provided invaluable help by describing the ETA algorithm detailedly, providing correct ETA descriptor values for some compounds, and taking part in the testing of the beta version.
- Version 2.9 [3 Dec 2011]: Updated CDK library to 1.4.6. This new version of CDK library has a bug fix which affects the value of chi valence descriptors (i.e. VCH-3, VCH-4, VCH-5, VCH-6, VCH-7, VC-3, VC-4, VC-5, VC-6, VPC-4, VPC-5, VPC-6, VP-0, VP-1, VP-2, VP-3, VP-4, VP-5, VP-6, VP-7) for all molecules containing SO2. In earlier versions of CDK library, the delta valence value for S in SO2 was calculated as 1.33 instead of 2.67. This was fixed in CDK 1.4.6. Thanks to Dr Tom Dickson for reporting the bug.
- Version 2.8 [2 Dec 2011]: Added variants of bond counts descriptors (nBonds2, nBondsS2, nBondsS3 and nBondsD2) and MAXDN, MAXDP, DELS descriptors as suggested by Prof Paola Gramatica and Dr Stefano Cassani. Added option to allow users to prevent automatic detection of aromaticity in molecules. This will enable users to have greater flexibility in deciding the aromaticity in molecular structures.
- Version 2.7 [8 Sep 2011]: Added feature to allow PaDEL-Descriptor to retain order of molecules in the descriptor file as suggested by Dr Tobias Kind. Added variants of hydrogen bond acceptor and donor counts descriptors (nHBAcc2, nHBAcc3, nHBAcc_Lipinski, nHBDon_Lipinski) as suggested by Prof Paola Gramatica and Dr Stefano Cassani. Added feature to allow PaDEL-Descriptor to calculate descriptors from a single structural file as suggested by Prof Alexander Tropsha and Dr Andy Fant. Added VABC descriptors. Added feature to allow PaDEL-Descriptor to get molecule name from filename instead of from within the file. Fixed bug with reading Hyperchem (HIN) files. Revert CDK library to 1.4.2.
- Version 2.6 [08 Jul 2011]: Change PubChem fingerprints (PubChemFP) to start numbering from 0 instead of 1 so as to be consistent with naming convention in PubChem fingerprints documentation as suggested by Prof Paola Gramatica.
- Version 2.5 [23 May 2011]: Added AcidicGroupCount, BasicGroupCount, FMF, and HybridizationRatio descriptors. Update CDK library to nightly build (15 May 2011) 1.5.0. Updated PaDEL-Descriptor to enable it to be used as a RapidMiner v5.1.6 extension.
- Version 2.4 [22 Sep 2010]: Fixed bug which prevents PaDEL-Descriptor from working properly. Added ability to write multiple molecules into a SDF file when used as a RapidMiner extension.
- Version 2.3 [29 Aug 2010]: Fixed bug which prevents PaDEL-Descriptor from working properly when used as a RapidMiner extension.
- Version 2.2 [29 Jun 2010]: Enable PaDEL-Descriptor to be used as a RapidMiner extension. Fixed bug in CDK which causes missing WeightedPathDescriptor descriptors when more than one thread is used. Fixed bug in reading PDB files.
- Version 2.1 [24 May 2010]: Change descriptor names from nB, nBs, nBd, nBt, nBq to bBonds, nBondsS, nBondsD, nBondsT, nBondsQ to prevent conflict in names between nB (number of bonds excluding bonds with hydrogen atoms) and nB (number of Boron atoms).
- Version 2.0 [17 May 2010]: Improved multi-thread capability. Added ring counts, count of chemical substructures identified by Laggner, and binary fingerprints and count of chemical substructures identified by Klekota and Roth. Added counts, minimum and maximum for atom type electrotopological state. Improved GUI to allow selection of descriptor and fingerprint types.
- Version 1.10 [03 Mar 2010]: Fixed bug which causes Java Exception errors if file to save descriptors to does not have a file extension.
- Version 1.9 [05 Jan 2010]: Added option to limit the maximum number of compounds to be saved to a descriptor file. Removed IPMolecularLearning as a default 2D descriptor to calculate as it is much slower to compute than other descriptors. Change Pubchem fingerprint to be the default fingerprint to be calculated.
- Version 1.8 [16 Dec 2009]: Fixed error where CPSADescriptor and PetitjeanShapeIndexDescriptor were wrongly classified as 2D descriptors. Fixed bug which does not show processing status for individual molecules in SDF files.
- Version 1.7 [04 Dec 2009]: Improve auto-detection of molecular file formats. Now file formats, except SMILES files, are auto-detected using CDK and does not depend on the extension of the file.
- Version 1.6 [03 Dec 2009]: Fixed bug which prevents the rest of the compounds from being processed if one compound has an error during processing. Added feature to automatically add csv extension to Descriptor output file if required.
- Version 1.5 [01 Dec 2009]: Reduce memory usage for large SDF, SMILES, PubChem ASN and PubChem XML files.
- Version 1.4 [26 Nov 2009]: Change GUI to using property sheet. Update CDK library to 1.3.1. Enable ALOGP, IPMolecularLearning descriptors, and ExtendedFingerprinter fingerprint. Added MannholdLogP descriptor and PubchemFingerprinter fingerprint.
- Version 1.3 [26 Mar 2009]: Fixed multi-thread bug which causes some descriptor values to be missing randomly.
- Version 1.2 [12 Feb 2009]: Fixed bug which prevents multiple compounds in a single SDF file from being processed. Update CDK library to 1.2.x branch.
- Version 1.1 [19 Sep 2008]: Removed TLSER descriptors and added MLFER and McGowanVolume descriptors.
- Version 1.0 [06 Aug 2008]: First release.
Last modified on 17 July, 2014 by Yap Chun Wei